Medical Devices: FDA Has Begun Building an Active Postmarket Surveillance System
Fast Facts
More than 1.7 million injuries and 83,000 deaths over a recent 10-year period in the U.S. have been potentially linked to faulty medical devices. These medical products range from surgical masks to implantable pacemakers.
FDA has begun building a surveillance system to look for potential safety issues in such devices. It plans to start with a few devices and expand over time. However, it has had problems getting funding and identifying patients who are using the devices. FDA is taking steps to overcome these challenges.
Oversight of the safety of medical products, including medical devices, has been a topic on our High Risk List since 2009.
Image of an implanted cardiac pacemaker
Highlights
What GAO Found
More than 1.7 million injuries and 83,000 deaths in the United States over a 10-year period were potentially linked to medical devices, according to a 2018 study of Food and Drug Administration (FDA) data. Medical devices include a wide range of products from surgical masks to implantable pacemakers. Active postmarket surveillance involves the ongoing review of evidence—derived from the analysis of data sources such as electronic health records, billing claims, pharmacy and other data—to detect medical device safety issues that may otherwise go unreported. FDA has taken steps to establish an active postmarket surveillance system for medical devices. These include:
- establishing a coordinating center in 2016 to partner with FDA to organize a network of data sources (health systems and other collaborators);
- completing in 2021 the cloud-based data infrastructure necessary to collect evidence of medical device performance while protecting patient privacy; and
- planning to begin active postmarket surveillance of two medical devices by December 2024, with plans to expand over 5 years (see figure).
Planned Expansion of FDA's Active Postmarket Surveillance System
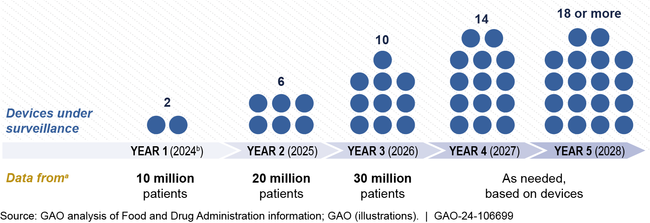
aRepresents anticipated patient data totals. Not all patients will have necessarily used the devices under surveillance.
bFDA anticipates completion of year 1 expansion by December 2024, contingent on funding availability.
FDA has faced two key challenges establishing its system, according to agency officials: (1) limited use of unique device identifiers in electronic health records and billing claims, which makes identifying devices used by patients more difficult; and (2) funding considerations to support active surveillance. FDA has taken actions to encourage use of unique device identifiers, such as coordinating with federal entities and publishing a document advertising the benefits of use to health systems. In addition, FDA has estimated current and future active surveillance costs and is considering options for how to fund the work by advocating for alternative funding sources.
GAO will continue to monitor FDA's progress in establishing an active postmarket surveillance system.
Why GAO Did This Study
FDA is responsible for ensuring the safety and effectiveness of medical devices marketed in the U.S. GAO has previously reported on challenges FDA has faced in its oversight of the safety of medical products, including medical devices, and designated this as a high-risk issue area since 2009. Federal law mandated in 2012 that FDA establish an active postmarket surveillance system for medical devices.
GAO was asked to review FDA's efforts related to postmarket surveillance of medical devices. This report identifies and discusses the steps FDA has taken to establish an active postmarket surveillance system, and the key challenges FDA has faced in establishing this system and actions it has taken to address them.
GAO reviewed documentation and interviewed officials from FDA and the coordinating center working with FDA to establish its active surveillance system. In addition, GAO interviewed representatives from three health systems and one research organization. These were selected in part based on the types of data they contributed to the network organized by the coordinating center. GAO also interviewed associations representing device manufacturers, health care providers, and patients for their views on FDA's efforts to establish its system. These were selected in part based on their work related to medical devices or active surveillance.
The Department of Health and Human Services (of which FDA is a part) and the coordinating center working with FDA provided technical comments on a draft of this report, which GAO incorporated as appropriate.
For more information, contact Mary Denigan-Macauley at (202) 512-7114 or DeniganMacauleyM@gao.gov.